Parallel programming
MPI
The MPI (Message Passing Interface) standard is an API for processes that need to send, wait or receive messages. A full documentation of all the implemented functions can be found in the MPI Standard documentation.
MPI implementations
The supercomputer comes with a default MPI implementation provided by the manufacturer, which is Open MPI supported as well as WI4MPI wrapper. These MPI stacks are strongly recommended and fully supervised by an expert team on the computing center. We ensure then end-to-end support including installation, configuration, performance optimization and issue resolution for e.g. scalability or debugging.
For other MPI libraries like MPICH or Intel MPI, only “best effort” support is provided to access them “as is” for ensuring availability, but without full guarantees on performance. It relies mainly on existing knowledge, documentation, and community support.
Other existing implementations include MVAPICH2 or Platform MPI but all are not made available on the cluster. To see a list of implementations available, use the command module avail mpi.
The default MPI implementation is loaded in your environment at connexion time.
Compiling MPI programs
You can compile and link MPI programs using the wrappers mpicc, mpic++, mpif77 and mpif90. Those wrappers actually call basic compilers but add the correct paths to MPI include files and linking options to MPI libraries.
For example to compile a simple program using MPI:
$ mpicc -o mpi_prog.exe mpi_prog.c
$ mpic++ -o mpi_prog.exe mpi_prog.cpp
$ mpif90 -o mpi_prog.exe mpi_prog.f90
To see the full compiling call made by the wrapper, use the command mpicc -show.
Wi4MPI
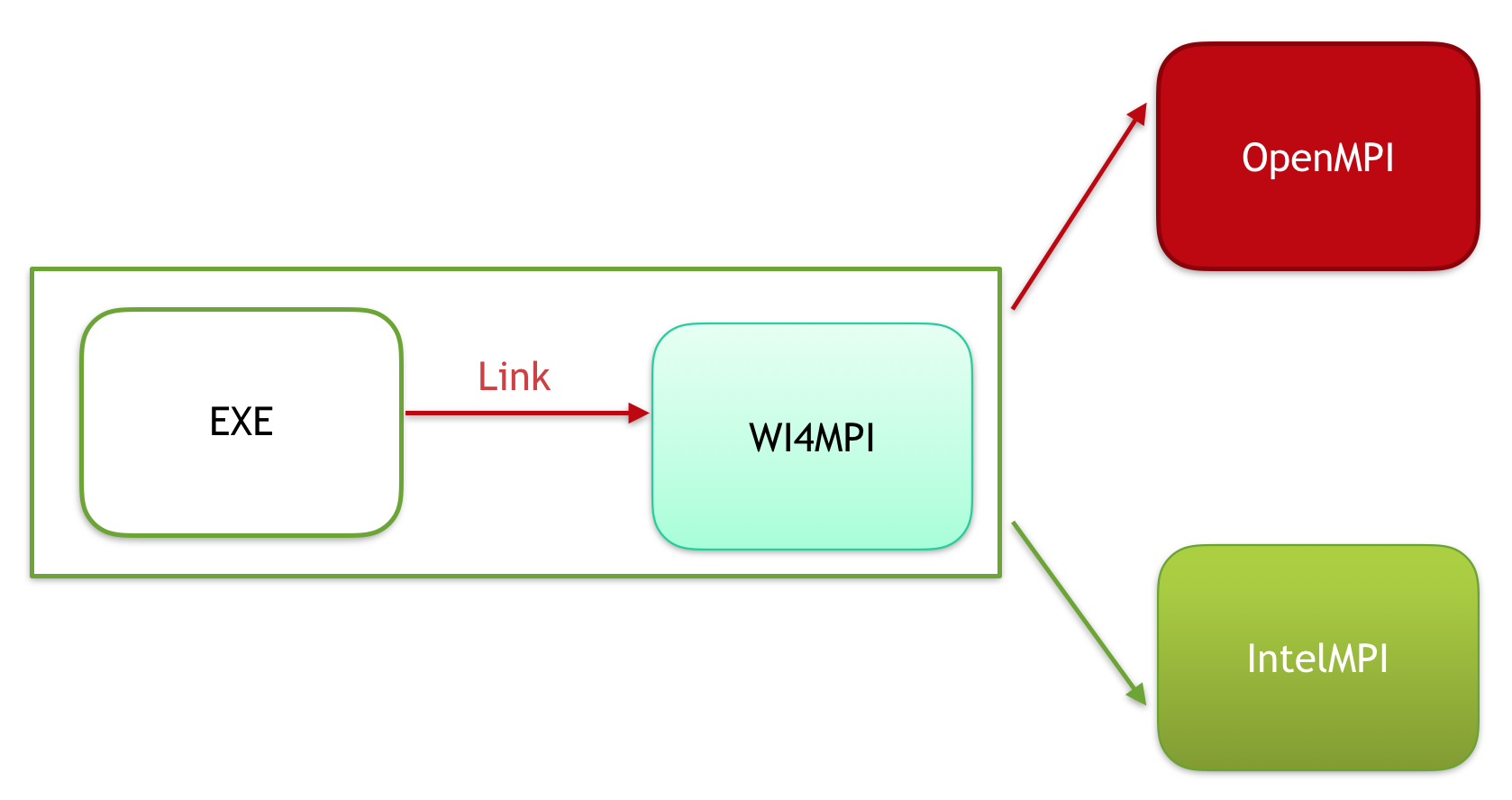
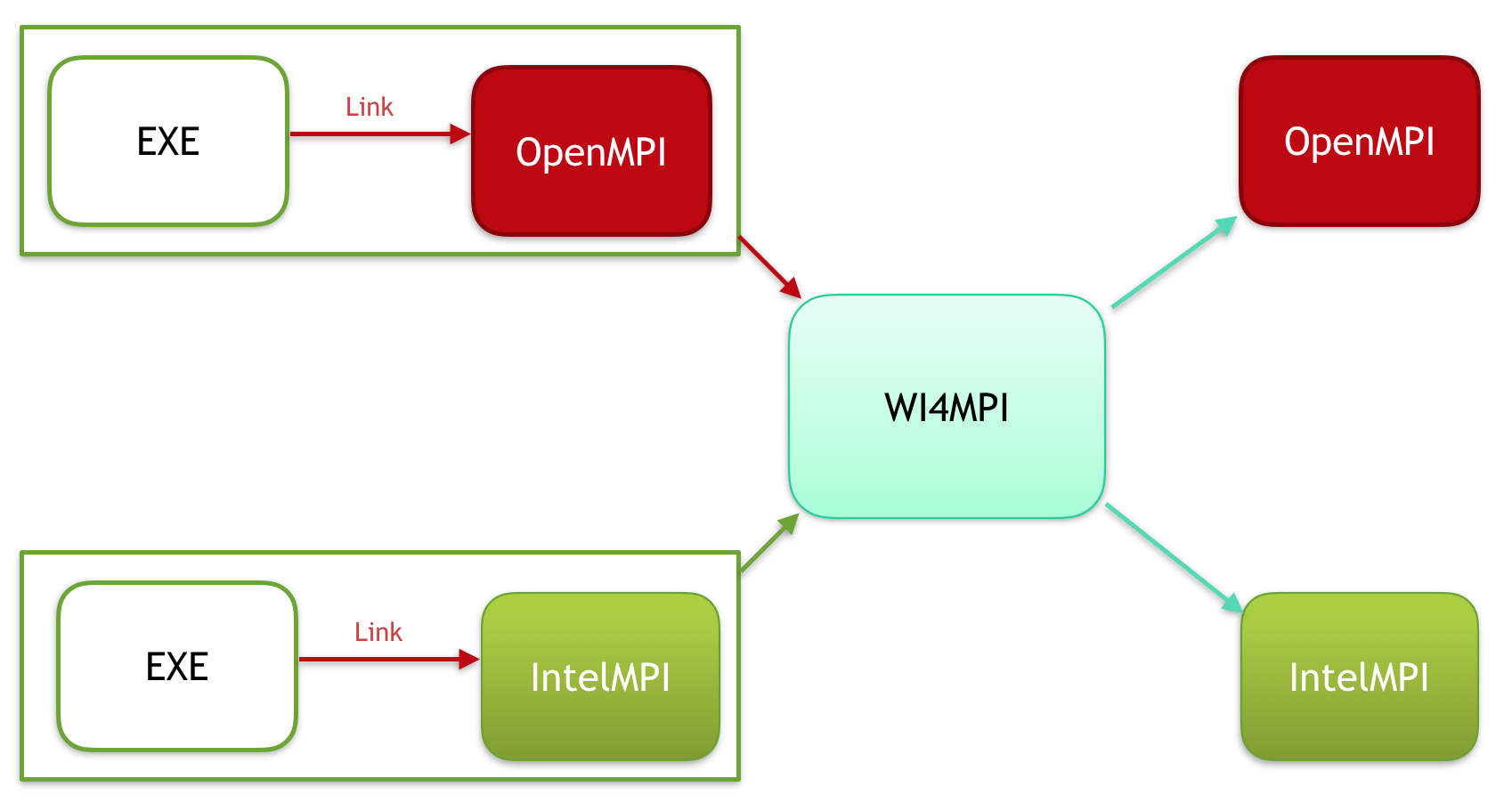
Interoperability between MPI implementations is usually not possible because each one has a specific Application Binary Interface (ABI). To overcome this, we have Wi4MPI. Wi4MPI provides two modes (Interface and Preload) with the same promise, one compilation for several run on different MPI implementations (OpenMPI, IntelMPI).
Interface
In this mode, Wi4MPI works as an MPI implementation.
$ module load mpi/wi4mpi
feature/wi4mpi/to/openmpi/x.y.z (WI4MPI feature to openmpi x.y.z)
wi4mpi/a.b.c (Wi4MPI with openmpi/x.y.z)
$ mpicc hello.c -o hello
$ ccc_mprun ./hello
By default, Wi4MPI is set to run application under OpenMPI. To choose the runtime MPI implementation please proceed as follow:
$ module switch feature/wi4mpi/to/openmpi/x.y.z feature/wi4mpi/to/intelmpi/a.b.c
interface
Preload
In this mode, Wi4MPI works as a plugin.
$ module load mpi/wi4mpi
feature/wi4mpi/to/openmpi/x.y.z (WI4MPI feature to openmpi x.y.z)
wi4mpi/a.b.c (Wi4MPI with openmpi/x.y.z)
This time the MPI implementation used for the compilation of the application needs to be provided as follow:
$ module load feature/wi4mpi/from/openmpi/x.y.z
$ module list
1) ccc 4) licsrv/intel 7) fortran/intel/x.y.z 10) mkl/x.y.z 13) hwloc/x.y.z 16) mpi/wi4mpi/x.y.z
2) datadir/own 5) c/intel/x.y.z 8) feature/mkl/lp64 11) intel/x.y.z 14) flavor/openmpi/cea 17) feature/wi4mpi/from/openmpi/x.y.z
3) dfldatadir/own 6) c++/intel/x.y.z 9) feature/mkl/sequential 12) flavor/wi4mpi/standard 15) feature/wi4mpi/to/openmpi/x.y.z
$ ccc_mprun exe_OpenMPI
To see all available features:
$ module avail feature/wi4mpi
To know more about Wi4MPI please visit the cea-hpc github.
Tuning MPI
OpenMPI
MCA Parameters
OpenMPI can be tuned with parameters. The command ompi_info -a gives you a list of all parameters and their description.
$ ompi_info -a
(...)
MCA mpi: parameter "mpi_show_mca_params" (current value: <none>, data source: default value)
Whether to show all MCA parameter values during MPI_INIT or not (good for
reproducibility of MPI jobs for debug purposes). Accepted values are all,
default, file, api, and environment - or a comma delimited combination of them
(...)
Theses parameters can be modified with environment variables set before the ccc_mprun command. The form of the corresponding environment variable is OMPI_MCA_xxxxx where xxxxx is the parameter.
#!/bin/bash
#MSUB -r MyJob_Para # Job name
#MSUB -n 32 # Number of tasks to use
#MSUB -T 1800 # Elapsed time limit in seconds
#MSUB -o example_%I.o # Standard output. %I is the job id
#MSUB -e example_%I.e # Error output. %I is the job id
#MSUB -q partition # Partition name
#MSUB -A <project> # Project ID
set -x
cd ${BRIDGE_MSUB_PWD}
export OMPI_MCA_mpi_show_mca_params=all
ccc_mprun ./a.out
For more information on MCA parameters, check out the tuning part of the openmpi FAQ.
Predefined MPI profiles
Some common MCA parameters are defined in several features (openmpi) to simplify their usage.
Here are those modules:
$ module avail feature/openmpi
----------------- /opt/Modules/default/modulefiles/applications -----------------
----------------- /opt/Modules/default/modulefiles/environment -----------------
feature/openmpi/big_collective_io feature/openmpi/gnu feature/openmpi/mxm feature/openmpi/performance feature/openmpi/performance_test
Here is their description and their limitation(s):
performance
Description : Improve communication performances in classic applications (namely those with fixed communication scheme). Limitation : Moderately increase the memory consumption of the MPI layer.
big_collective_io
Description : Increase data bandwidth when accessing big files on lustre file system. Limitation : Only useful when manipulating big files through MPI_IO and derivated (parallel hdf5, etc.).
collective_performance
Description : Improve the performance of several MPI collective routines by using the GHC feature developed by BULL. Limitation : Improvements may not be seen on small cases. Namely change the order in which MPI reduction operations are performed and may impact the reproductability and/or the numerical stability of very sensible systems.
low_memory_footprint
Description : Reduce the memory consumption of the MPI layer. Limitation : May have strong impact over communication performances. Should only be used when you are near the memory limits.
Compiler Features
By default, MPI wrappers use Intel compilers. To use GNU compilers, you need to set the following environment variables:
OMPI_CCfor COMPI_CXXfor C++OMPI_F77for fortran77OMPI_FCfor fortran90
For example:
$ export OMPI_CC=gcc
$ mpicc -o test.exe test.c
It’s also possible to use the feature feature/openmpi/gnu which set all variables define above at GNU compilers (gcc, g++, gfortran).
$ module load feature/openmpi/gnu
load module feature/openmpi/gnu (OpenMPI profile feature)
IntelMPI
Compiler Features
By default, MPI wrappers use Intel compilers. To use GNU compilers, you need to set the following environment variables:
I_MPI_CCfor CI_MPI_CXXfor C++I_MPI_F77for fortran77I_MPI_FCfor fortran90
For example:
$ export I_MPI_CC=gcc
$ mpicc -o test.exe test.c
It’s also possible to use the feature feature/intelmpi/gnu which set all variables define above at GNU compilers (gcc, g++, gfortran).
$ module load feature/intelmpi/gnu
load module feature/intelmpi/gnu (IntelMPI profile feature)
OpenMP
OpenMP is an API that supports multi-platform shared memory multiprocessing programming in C, C++, and Fortran. It consists of a set of compiler directives, library routines, and environment variables that influence run-time behavior. More information and a full documentation can be found on the official website.
Compiling OpenMP programs
The Intel and GNU compilers both support OpenMP. Use the -fopenmp flag to generate multi-threaded code with those compilers.
Note
-openmp works only for intel whereas -fopenmp works with both Intel and GNU compilers.
$ icc -fopenmp -o openmp_prog.exe openmp_prog.c
$ icpc -fopenmp -o openmp_prog.exe openmp_prog.cpp
$ ifort -fopenmp -o openmp_prog.exe openmp_prog.f90
Intel thread affinity
By default, threads inherit the same affinity than their parent process (see Process distribution, affinity and binding for more information). For example, if the parent process has been allocated 4 cores, the OpenMP threads spawned by this process will be allowed to run freely on those 4 cores.
To set a more specific binding for threads among the allocated cores, Intel provides environment variables: KMP_AFFINITY
The values given to KMP_AFFINITY should be a comma-separated list of the following keywords:
verbose: prints messages concerning the supported affinity.granularity=: specifies whether to pin OpenMP threads to physical cores (granularity=core, this is the default) or pin to logical cores (granularity=fine). This is only effective on nodes that support SMT (Simultaneous Multithreading, as hyperthreading on Intel architecture for instance).compact: assigns the threads as close as possible together.scatter: distributes the threads as evenly as possible across the entire system.
GPU-accelerated computing
This section is about GPU-accelerated compute jobs. For information about GPU-accelerated remote desktop visualization service, please refer to the Interactive access section.
CUDA
CUDA may refer to
The CUDA Language Extentions: A set of language extentions for C, C++ and Fortran.
The CUDA Driver: A low level API for interacting with GPU devices.
The CUDA Runtime API: A higher-level programming interface for developping GPU-accelerated code.
The CUDA Runtime library: The implementation of the CUDA Runtime API
The CUDA Toolkit: A set of tools and libraries distributed by NVIDIA including the CUDA runtime Libraries.
NVIDIA graphics cards are operated through the CUDA driver. It is a low level API offering fine-grained control which requires explicit management of cuda code, CUDA module loading, and execution context. The CUDA runtime API is a higher level, more easy-to-use programming interface. Most applications do not use the driver API as they do not need the finer level of control. The CUDA runtime API provides all functions required for running GPU computations on NVIDIA graphics cards such as device management, thread management, memory allocation and copying, event management and execution control. Visit the online CUDA documentation for more informations.
The CUDA programming model includes a number of language extentions to C and C++. CUDA Fortran directive-based compiler supports similar functionnalities. OpenMP and OpenACC are other programming models that can be used for writing efficient GPU-accelerated programs and are supported by the NVIDIA and GCC compilers.
Overview of NVHPC
The NVHPC toolkit includes a number of compilers and libraries designed for GPU programming. It is available though the nvhpc module :
$ module load nvhpc
NVHPC includes the nvc, nvfortran, nvc++ and nvcc compilers.
nvc
nvc is a C11 compiler for NVIDIA GPUs and AMD, Intel, OpenPOWER, and Arm CPUs. It invokes the C compiler, assembler, and linker for the target processors with options derived from its command line arguments. nvc supports ISO C11, supports GPU programming with OpenACC, and supports multicore CPU programming with OpenACC and OpenMP.
nvc++
nvc++ is a C++17 compiler for NVIDIA GPUs and AMD, Intel, OpenPOWER, and Arm CPUs. It invokes the C++ compiler, assembler, and linker for the target processors with options derived from its command line arguments. nvc++ supports ISO C++17, supports GPU and multicore CPU programming with C++17 parallel algorithms, OpenACC, and OpenMP.
nvfortran
nvfortran is a Fortran compiler for NVIDIA GPUs and AMD, Intel, OpenPOWER, and Arm CPUs. It invokes the Fortran compiler, assembler, and linker for the target processors with options derived from its command line arguments. nvfortran supports ISO Fortran 2003 and many features of ISO Fortran 2008, supports GPU programming with CUDA Fortran, and GPU and multicore CPU programming with ISO Fortran parallel language features, OpenACC, and OpenMP.
nvcc
nvcc is a CUDA C/C++ compiler driver for NVIDIA GPUs. nvcc accepts a range of conventional compiler options, such as for defining macros and include/library paths, and for steering the compilation process. All non-CUDA compilation steps are forwarded to a C++ host compiler.
NVHPC also comes with a set of mathematic libraries such as cublas, cufft, and nccl.
CUDA Math API
The CUDA math API supports the usual intrinsics and mathematical functions for integer, 64-bit, 32-bit and 16-bit floats.
cuBLAS
The cuBLAS library is an implementation of BLAS (Basic Linear Algebra Subprograms) on top of the NVIDIA CUDA runtime. It allows the user to access the computational resources of NVIDIA Graphical Processing Unit (GPU), but does not auto-parallelize across multiple GPUs.
cuSPARSE
cuSPARSE implements linear algebra for sparse vectors and matrices. It supports coo, csr and csc sparse formats.
cuSOLVER
Based on the cuBLAS and cuSPARSE libraries, cuSOLVER implements two high-level API for dense and sparse linear algebra. The cuSolver API provides single-GPU LAPACK-like features. The cuSolverMG API provides an implementation of the ScaLAPACK API for single node multi-GPU execution.
NVBLAS
NVBLAS is another implementation of most level 3 BLAS routines. It supports dynamic execution on multiple GPUs. It is built on top of the cuBLAS library.
cuFFT
The CUDA Fast Fourier Transform Library supports batched, multi-GPU, 1D, 2D and 3D transforms. Il implements and extends the FFTW3 API.
NCCL
The NVIDIA Collective Communications Library (NCCL, pronounced “Nickel”) is a library providing inter-GPU communication primitives that are topology-aware and can be easily integrated into applications.
NCCL implements both collective communication and point-to-point send/receive primitives. It is not a full-blown parallel programming framework; rather, it is a library focused on accelerating inter-GPU communication.
The documentation for all of these can be found at nvidia.com: NVIDIA HPC Documentation
Using NVHPC libraries
nvc, nvcc and nvfortran compilers accept special compilation flags in order to enable compilation with these libraries using the following syntax, which will use the libaries available from -L command line arguments or from LD_LIBRARY_PATH
-cudalib[=cublas|cufft:{callback}|cufftw|curand|cusolver|cusparse|cutensor|nvblas|nccl|nvshmem|nvlamath]
In order to compile and link your code using the libraries included in nvhpc, you need to load nvhpc and/or its dependencies so that the required libraries are available from LD_LIBRARY_PATH
$ module load nvhpc
$ module show math/nvidia
[...]
append-path LD_LIBRARY_PATH /ccc/products/nvhpc-21.2/system/default/Linux_x86_64/21.2/math_libs/lib64
[...]
setenv MATH_NVIDIA_LIBDIR /ccc/products/nvhpc-21.2/system/default/Linux_x86_64/21.2/math_libs/lib64
[...]
$ ls $MATH_NVIDIA_LIBDIR
libcublas.so libcublasLt.so libcublasLt.so.11
libcublasLt.so.11.4.2.10064 libcublasLt_static.a
libcublas.so.11 libcublas.so.11.4.2.10064
libcublas_static.a libcufft.so
libcufftw.so libcufftw.so.10 libcufftw.so.10.4.2.58
libcufftw_static.a libcufft.so.10
libcufft.so.10.4.2.58 libcufft_static.a
libcufft_static_nocallback.a libcurand.so
libcurand.so.10 libcurand.so.10.2.4.58 libcurand_static.a
[...]
If you want to use some libraries included in NVHPC that are not direcly available using such environment variables, use paths relative to variables such as NVHPC_ROOT or MATH_NVIDIA_ROOT
$ NVSHMEM_LIBDIR=$MATH_NVIDIA_ROOT/../comm_libs/nvshmem/lib/
$ LD_LIBRARY_PATH=$LD_LIBARY_PATH:$NVSHMEM_LIBDIR
You may also use -I or -L compilation and linking options to specify the paths of includes and libraries directories to your favorite C/C++ compiler.
Using NVCC
NVCC is a helper driver for compiling CUDA C/C++ code. It handles a number source code transformation, invokes the underlying CUDA compiler as well as a general purpose C++ host compiler (g++ by default) for compiling non-CUDA code and preprocessing. Its purpose is to use the development of CUDA applications.
$ module load nvhpc
$ nvcc -o test_cuda test.cu
GPU Hardware Architectures, and the set of features supported by a particular iteration over a general architecture are identified by the compute capabilities of a target GPU. As such, compute capabilities 7 refer to the Volta architecture while compute capabilities 8 refer to the Amper architecture.
Use the following command to list the GPU architectures as well as the virtual device architectures supported by the NVCC compiler.
$ nvcc --list-gpu-code --list-gpu-arch
Use the -gencode argument to NVCC to specify the target GPU architecture as well as the virtual device architecture.
You may specify several levels of support if you need the code to run on different GPU generations.
For generating GPU code for both V100 and A100 GPUs, use
$ nvcc -gencode arch=compute_72,code=sm_72 -gencode arch=compute_80,code=sm_80 -o test_cuda test.cu
The nvcc command uses C and C++ compilers underneath. Compiling with nvcc -v will show the detail of the underlying compiling calls. By default, the GNU compilers are used. To change the underlying compiler, use the -ccbin option:
$ module load nvhpc
$ module load intel
$ nvcc -v -ccbin=icc -o test_cuda test.cu
Most GPU codes are partially composed of basic sequential codes in separate files. They may be compiled separately:
$ ls
cuda_file.cu file.c
$ icc -o file.o -c file.c
$ nvcc -ccbin=icc -o cuda_file.o -c cuda_file.cu
$ icc -o test_cuda -L${CUDA_LIBDIR} -lcudart file.o cuda_file.o
CUDA fortran
The CUDA Fortran language extentions allow programming for GPU in Fortran using similar idioms to C/C++ CUDA.
To compile CUDA Fortran code, use the nvfortran compiler as described in section Compiling OpenACC and OpenMP code for GPU with the -cuda option in place of the -acc option.
Compiling OpenACC and OpenMP code for GPU
Based on the LLVM compiler, nvc, nvc++ and nvfortran can be used for compiling C, C++ and Fortran code enriched with OpenMP or OpenACC directives gor execution on NVIDIA GPUs.
They determine the source input type by examining the filename extentions, and use the following conventions:
- fortran sources
- filename.f
indicates a Fortran source file.
- filename.F
indicates a Fortran source file that can contain macros and preprocessor directives (to be preprocessed).
- filename.FOR
indicates a Fortran source file that can contain macros and preprocessor directives (to be preprocessed).
- filename.F90
indicates a Fortran 90/95 source file that can contain macros and preprocessor directives (to be preprocessed).
- filename.F95
indicates a Fortran 90/95 source file that can contain macros and preprocessor directives (to be preprocessed).
- filename.f90
indicates a Fortran 90/95 source file that is in freeform format.
- filename.f95
indicates a Fortran 90/95 source file that is in freeform format.
- filename.cuf
indicates a Fortran 90/95 source file in free format with CUDA Fortran extensions.
- filename.CUF
indicates a Fortran 90/95 source file in free format with CUDA Fortran extensions and that can contain macros and preprocessor directives (to be preprocessed).
- C/C++ sources
- filename.c
indicates a C source file that can contain macros and preprocessor directives (to be preprocessed).
- filename.C
indicates a C++ source file that can contain macros and preprocessor directives (to be preprocessed).
- filename.i
indicates a preprocessed C or C++ source file.
- filename.cc
indicates a C++ source file that can contain macros and preprocessor directives (to be preprocessed).
- filename.cpp
indicates a C++ source file that can contain macros and preprocessor directives (to be preprocessed).
Here is an example of compilation using the nvfortran compiler, highlighting a few useful compilation options:
$ nvfortran -acc=gpu,host -gpu=cc70,cc80,time -Minfo=accel -fast -c src/example_file.f90 -o bin/example_file.o -module bin
$ nvfortran -acc=gpu,host -gpu=cc70,cc80,time -Minfo=accel -fast -o bin/example_bin bin/example_file.o ccc_mprun ./example_bin
-acc=gpu,hostenables OpenACC directives for execution on GPU when at least one such device is available, or sequantial CPU execution otherwise.
-mp=gpuenables OpenMP directives for execution on GPU devices.
-mp=multicoreenables OpenMP directives for execution on CPU.
-gpu=cc70,cc80specifies that both V100 and A100 GPU architectures should be targeted for GPU executable code.
-gpu=timeenables light profiling of the accelerator regions and generated kernels. A profiling report is printed at the end of the resulting program execution. This can also be achieved by setting the environment variable
NVCOMPILER_ACC_TIMEto a non-zero value during compilation.-Minfo=accelenables output of information about accelerator compute kernel compilation. Use
-Minfo=allfor an even more verbose compilation output, including general and loop-specific optimization information.-fastenables the most impactful optimization options. Use
-O2or lower optimization level instead for development phases since-fastdisables stack flame generation and makes execution analysis and debugging generally more complex.
Other useful preprocessing, compiling and linking options such as -Mfree (assume free-format fortran source), are available. Please refer to man <compiler>, <compiler> --help, or NVIDIA’s HPC Compilers User’s Guide for more information
Compiling GPU-accelerated MPI programs
The mpi/openmpi module is available with NVHPC compilers support.
Loading the following sequence of modules enables using the mpicc, mpic++ and mpif90 compiler wrappers with the NVHPC compilers:
$ module load nvhpc
$ module load mpi/openmpi
You may then use mpif90 in place of nvfortran for compiling and linking distributed, GPU-accelerated mpi applications.
Accelerated MPI routines are available for transfering data between GPUs inside and across nodes.
Specific configurations are loaded when loading MPI with nvhpc on GPU partitions to enable GPU Direct RDMA, Device to Device, and CUDA-aware MPI, enabling specific programming practices such as initiating MPI communications using device pointers in cuda, or use of the host data use device OpenACC directive.
Running GPU applications
This section is about GPU-accelerated compute jobs. For information about GPU-accelerated remote desktop visualization service, please refer to the Interactive access section. Programs compiled with cuda are started normally and will use the cuda runtime libraries and drivers in order to offload computations to the GPUs. GPU MPI programs are started normally using ccc_mprun.
Simple one GPU job
#!/bin/bash
#MSUB -r GPU_Job # Job name
#MSUB -n 1 # Total number of tasks to use
#MSUB -c 32 # Assuming compute nodes have 128 cores and 4 GPUs
#MSUB -T 1800 # Elapsed time limit in seconds
#MSUB -o example_%I.o # Standard output. %I is the job id
#MSUB -e example_%I.e # Error output. %I is the job id
#MSUB -q <partition> # Partition name
#MSUB -A <project> # Project ID
set -x
cd ${BRIDGE_MSUB_PWD}
module load nvhpc
ccc_mprun ./a.out
Multi GPUs job
Use the command ccc_mpinfo under the section “GpN” to aknowledge the number of GPUs per node.
Assuming “GpN” GPUs per node, each GPU reservation allocates 1/ “GpN” th of a node. When all “GpN” GPUs are reserved, a full node is allocated. For job allocations using more than one full node, additionnal nodes are fully allocated. (see Process distribution, affinity and binding).
Use the #MSUB -c option to tune the number of cores per task.
Assuming a node has 128 CPU cores and 4 GPUs, here is non-exhaustive list of examples:
-n 1 -c 1option doesn’t allocate any GPUs
-n 1 -c 32option allocates 1 GPU per task
-n 1 -c 33option allocates 1 GPU per task
-n 1 -c 64option allocates 2 GPUs per task
-n 1 -c 96option allocates 3 GPUs per task
-n 1 -c 128option allocates 4 GPUs per task
-n 4 -c 32option allocates 1 GPUs per task and 4 total GPUs for the job
-n 8 -c 32option allocates 1 GPUs per task and 8 total GPUs for the job in two full nodes
Note
To ensure full GPU allocation, use a factor n of CpN/GpN to allocate n GPUs per task (i.e. for a partition with CpN=128 and GpN=4, CpN/GpN=32). CpN stands for CPU cores per Node and can be aknowledged with the command ccc_mpinfo.
The --gpu-bind=closest Slurm option binds each task to the GPUs and CPU cores which are closest. It is equivalent to the --tres-bind=gres/gpu:closest option.
For example, in order to use 4 task per node on 2 nodes, each task managing a single GPU, you may use the following job script.
#!/bin/bash
#MSUB -r MPI_GPU_Job # Job name
#MSUB -n 8 # Total number of tasks to use
#MSUB -c 32 # Number of CPU cores per task
#MSUB -T 1800 # Elapsed time limit in seconds
#MSUB -o example_%I.o # Standard output. %I is the job id
#MSUB -e example_%I.e # Error output. %I is the job id
#MSUB -q <partition> # Partition name
#MSUB -A <project> # Project ID
#MSUB -E "--gpu-bind=closest" # Bind each task to the closest GPU
set -x
cd ${BRIDGE_MSUB_PWD}
module load nvhpc mpi
ccc_mprun ./myprogram
This example implementation associates each GPU with the tasks according to the NUMA topology of the machine. For further informations, refer to the output of the nvidia-smi topo -m command to check the actual NUMA affinity of the CPU cores for each nvidia graphics device.
For more general information about tasks placement and binding, please refer to the Process distribution, affinity and binding section.
Debugging, profiling and performance analysis
nvidia-smi
nvidia-smi is a useful monitoring and management tool. It shows the available GPUs, running processes, memory usage. Use
nvidia-smi topo -mto display GPUDirect interconnection topology.Note that repeatedly polling GPU usage through repeated calls to nvidia-smi or usage of the
--loopor--loop-msoptions may impact performance.The
dmonanddaemonoptions may help setting up monitoring of GPUs usage.
CUDA-GDB
CUDA-GDB is an extention to gdb for debugging C/C++ and Fortran CUDA applications running on actual hardware. It supports program stepping, breakpoints, thread and memory inspection for CUDA code in addition to the usual gdb capabilities regarding CPU code.
CUPTI
CUPTI is the CUDA Profiling Tools Interface. It enables the creation of profiling and tracing tools that target CUDA applications. Its documentation can be found online.
Nsight Systems
Please refer to the the dedicated section.
Nsight Compute
Please refer to the the dedicated section.